En el
contexto de este capítulo Vd será invitado
también a visitar estos apartados...
El
análisis e interpretación de la
función de densidad
electrónica, es decir, la
resolución de una estructura cristalina (molecular, o no
molecular), nos conduce a un modelo inicial, no
definitivo, que se
describe por las
posiciones relativas de los átomos, los cuales pueden
representarse
mediante puntos o pequeñas esferas:



Izquierda:
Modelo
inicial de la estructura
tridimensional de una molécula.
Los átomos están representados por
pequeñas
esferas.
Centro: Modelo
inicial de la estructura tridimensional de una molécula. A
pesar
de la mayor o menor belleza del modelo, la
cantidad de información
es prácticamente la misma que en el modelo de la izquierda.
Derecha: Modelo
inicial de la estructura tridimensional de una molécula,
incluyendo la estructura cristalina (empaquetamiento). La
visión muestra los ejes de la celdilla
elemental.
Pero
una vez conseguido el modelo estructural completo, con
total
sentido estereoquímico e incluyendo el empaquetamiento
cristalino, es necesario sacar el máximo
partido a las medidas experimentales, ya que, en general, se dispone de
una cierta sobredeterminación de la información
experimental (patrón de difracción).
Por
ejemplo, para una estructura de tamaño mediano, con unos 50
átomos independientes (en la unidad asimétrica,
es decir,
en la parte que se repite por las operaciones de simetría)
normalmente se dispone de hasta aproximadamente 2.500
factores
de estructura, lo que
supone disponer de hasta 50
observaciones por
átomo. Este superávit de información
experimental normalmente no llega hasta estos límites en el
caso de
estructuras
más complejas, tal como ocurre en el caso de las
macromoléculas.
AJUSTE
FINAL DEL MODELO
Los parámetros fundamentales asociados a cada
átomo en
una estructura tridimensional son, obviamente, las tres coordenadas (x,
y,
z)
posicionales de cada átomo referidas a los ejes del sistema
de referencia (celdilla elemental).
Pero, en general, dada la sobredeterminación experimental
que en
general proporciona la difracción, el modelo
cristalográfico puede permitirse
ampliar
su complejidad, asociando además a cada átomo un parámetro
"térmico" que da cuenta de su estado de
vibración térmica
isotrópica (esférica) alrededor de su
posición de equilibrio y que se vería
representado por el mayor o menor volumen de las esferas de la
representación gráfica. Por lo tanto el modelo
isotrópico está representado por 4 variables:
3 posicionales + 1 térmica (que equivale al radio de la
esfera de vibración).
Sin embargo, en el caso de estructuras pequeñas y medianas
(hasta algunos centenares de átomos), el experimento de la
difracción suele dar de sí para poder completar
algo más el modelo de vibración
térmica de cada átomo, asignando un tensor (6
variables) a cada posición atómica, que
expresa
el estado de vibración del átomo de un modo
anisotrópico,
es decir, distinguiendo entre diferentes direcciones de
vibración en
forma de elipsoide (que
se asemeja a la forma de una pelota de "base-ball"). De este modo, el
modelo
cristalográfico anisotrópico requiere 9
variables por cada átomo (3 posicionales + 6 vibracionales).


Izquierda: Representación
gráfica de tres
átomos enlazados mediante los modelos de
vibración termica
isotrópica
Derecha: Los
mismos átomos de la figura de la izquierda, pero
representados mediante el modelo de vibración
termica
anisotrópica

 Izquierda: Modelo
anisotrópico de la
estructura tridimensional de una molécula y de fragmentos de
sus vecinas.
Derecha: Modelo
anisotrópico de la
estructura tridimensional de una molécula y su
empaquetamiento en el cristal.
Izquierda: Modelo
anisotrópico de la
estructura tridimensional de una molécula y de fragmentos de
sus vecinas.
Derecha: Modelo
anisotrópico de la
estructura tridimensional de una molécula y su
empaquetamiento en el cristal.
Independientemente del modelo isotrópico o
anisotrópico, y debido a la sobreabundancia de datos
experimentales de que, normalmente, se dispone a través del
experimento de difracción, el resultado
cristalográfico tiene capacidad para evolucionar hasta
valores posicionales y de vibración térmica muy
precisos que redundan en la precisión de cualquier
parámetro geométrico que se derive de la
estructura (distancias interatómicas, ángulos de
enlace, etc.).
La obtención de este modelo "ajustado" (afinado),
isotrópico o anisotrópico, es consecuencia de
metodología matemática analítica como
la de los mínimos cuadrados. Mediante esta
técnica se "mueven" ligeramente las posiciones
atómicas (las coordenadas) y se aplican factores
térmicos a cada átomo de tal modo que el
patrón
de difracción calculado con dicho modelo sea
prácticamente igual que el experimental (observado), es
decir
que se minimizan las diferencias entre los factores de estructura
observados y calculados. Este
proceso se lleva a cabo minimizando (haciendo tender a cero) la
función:
Σ
w |
|Fo| - |Fc|
|2
→ 0
Función de mínimos
cuadrados que se utiliza para ajustar el modelo final de las estructuras
en
donde w
representa un factor de "peso" asignado a cada observación,
separando así los efectos de aquellas observaciones
más precisas de las menos precisas y evitando así
errores sistemáticos en las observaciones experimentales que
pudieran sesgar el modelo.
Fo
y Fc
representan los factores de estructura observados y calculados,
respectivamente.
El ajuste consiste en modificar ligeramente las coordenadas de los
átomos del modelo para conseguir que los factores de
estructura
calculados (Fc)
con dicho modelo se parezcan los más posible a los factores
de estructura observados en el experimento de difracción (Fo).
Aunque la sobredeterminación experimental mencionada suele
asegurar el buen término de este proceso
matemático de ajuste, éste debe de ser controlado
con sentido estereoquímico, es decir, comprobando que los
pequeños movimientos posicionales de los átomos
sean razonables y que por lo tanto generan distancias
interatómicas dentro de lo esperable. Del mismo modo, los
factores de vibración térmicos
(isotrópicos ó anisotrópicos)
asociados a los
átomos deben de mostrar valores razonables.
Además del control aludido de los ligeros cambios que
sufre el modelo durante este proceso de ajuste, parece obvio
que (si todo va bien), además, el patrón de
difracción calculado (Fc) con
el modelo (coordenadas + factores de vibración)
explicará cada
vez mejor el patrón de difracción observado (Fo). La
comparación entre ambos patrones (observado vs. calculado) se
realiza mediante el denominado parámetro R,
que define el factor de "desacuerdo" entre ambos patrones:
R =
Σ [
| |Fo| - |Fc|
| ] / |Fo|
Factor
de desacuerdo del modelo
estructural, estimado en función de las diferencias entre
los
factores de estructura observados y los calculados con el modelo final
El valor del factor de desacuerdo (R)
suele estimarse en forma porcentual (%), es decir, multiplicado por
100, de tal modo que estructuras bien resueltas y con grado de
precisión adecuado muestran un factor R
siempre
por debajo de 0.10 (10%), lo cual implica que el
patrón de
difracción calculado difiere del observado (experimental)
menos
del 10%.
En las estructuras de las macromoléculas (enzimas,
proteínas, etc.), es normal que no exista una
sobredeterminación de valores experimentales y por lo tanto
dificilmente se llegan a usar un modelos estructurales
anisotrópicos. En estos casos, además, los
valores de desacuerdo (R)
son mayores que en el caso de las moléculas
pequeñas y medianas, de tal modo que valores del orden del
20%,
o algo menores, suelen ser aceptables. Además, como
consecuencia
de esta
escasez relativa de datos experimentales, el procedimiento
analítico de ajuste mencionado (mínimos
cuadrados) debe
de combinarse con un proceso de modelado
estereoquímico
interactivo e imponiendo ciertas "restricciones suaves" a la
geometría de la molécula.
VALIDACIÓN DEL MODELO
Existen una serie de herramientas que ayudan
a evaluar la fiabilidad de
un modelo estructural, y que en términos
cristalográficos se conocen
con el nombre de
validación,
de tal modo que
el modelo estructural obtenido
debe ser
contínuamente contrastado y validado
mediante criterios estereoquímicos consistentes. Es
decir, las distancias interatómicas y ángulos de
enlace deben ser aceptables. No lo sería, por
ejemplo, una distancia C---O de 0.8 Angström para un supuesto
grupo carbonilo (C=O). Y del mismo modo, los ángulos de
enlace, deben de ser consistentes con una geometría
aceptable. Estos criterios son muy restrictivos para los modelos
estructurales de compuestos de hasta mediana complejidad, pero incluso
en las estructuras de las macromoléculas deben de cumplirse
unos mínimos
Dispersión máxima
aceptable para distancias interatómicas y ángulos
de enlace en un modelo estructural de una macromolécula
Igualmente,
en el caso de las proteínas, debido al
carácter geométrico del
enlace peptídico (enlace entre dos aminoácidos
consecutivos), debe de cumplirse que el ángulo de
torsión en dicho enlace no debe desviarse mucho del
valor aceptable para la conformación estructural que adoptan
esos aminoácidos, tal como se muestra en la denominada
distribución de Ramachandran:
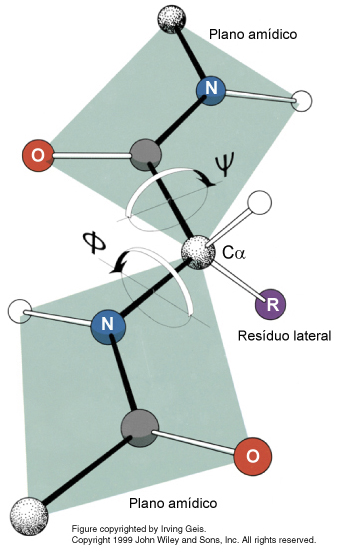

Izquierda: Esquema
del enlace peptídico,
mostrando los dos ángulos de torsión (Ψ y Φ) que
lo
definen. Pueden observarse también estos ángulos pinchando
en este enlace.
Derecha: Plot de
Ramachandran: Zonas
de dispersión aceptable para los valores de los
ángulos de torsión de
los enlaces peptídicos en un modelo estructural de una
macromolécula, dependiendo de las diferentes zonas
estructurales (alfa-hélices, láminas-beta, etc.)
Del mismo modo, los valores de los factores térmicos
asociados a cada átomo deben corresponder a valores
físicamente aceptables. Estos parámetros
vibracionales dan cuenta de la movilidad de diferentes zonas
estructurales. Así, en la estructura de una
macromolécula, éstos valores deben ser coherentes
con las zonas internas o externas de la molécula, siendo en
general menores para el interior, y mayores para las zonas externas,
próximas al solvente.

Representación
gráfica de la cadena principal de una
macromolécula en
donde los colores muestran los factores de
vibración
térmica. Colores "fríos"
(azules)
denotan zonas de escasa movilidad. Colores "calientes" (verdes
y rojos)
denotan zonas mucho más móviles.
GRADO
DE FIABILIDAD DEL
MODELO
Un modelo que ha sido
"validado" de acuerdo con los criterios descritos, es decir, que
muestra:
- acuerdo razonable entre los módulos de los
factores de estructura observados (experimentales) y calculados,
- distancias interatómicas, ángulos de
enlace y
ángulos de
torsión aceptables, es decir que cumple
con criterios
esteroquímicos aceptables, y
- factores de vibración térmica
físicamente razonables,
es un modelo fiable. Sin embargo, el concepto de fiabilidad no es un
parámetro totalmente cuantizable o expresable mediante un
único número. Por lo tanto, para interpretar un
modelo
estructural
hasta sus últimas consecuencias hay que tener en cuenta
que éste es una representación simplificada,
construida
sobre una función de densidad electrónica:
Pero
en cualquiera de los casos, el cristalógrafo llega
a
parámetros
atómicos (posicionales y vibracionales) con su
correspondiente valor de precisión. Este hecho provoca que
cualquier parámetro
cristalográfico directo (coordenadas
atómicas y factores de vibración) o indirecto
(distancias, ángulos, etc.) se exprese como un valor
numérico seguido de otro (entre paréntesis) que
expresa la denominada desviación estándard del
anterior y que afecta a la última cifra del valor expresado.
Por ejemplo, una distancia interatómica expresada en
Angströms como 1.541 (2) implica un valor numérico
de 1.541 y una desviación estándard de 0.002.
LA CONFIGURACIÓN (O ESTEREOQUIMICA) ABSOLUTA
Ya se ha comentado en un apartado anterior
que en todas aquellas estructuras en las que no existen planos de
simetría ni centros de inversión se puede hablar
de "configuración
absoluta", es decir que la estructura en cuestión
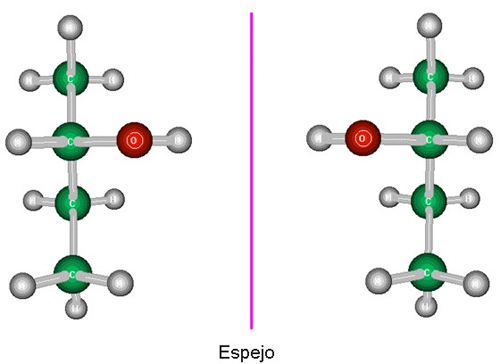
es diferente de su imagen especular:
Modelos estructurales de dos
moléculas enantiómeras entre sí (una
es imagen especular de
la
otra)
Pues bien, este matiz o diferencia estructural, que es de vital
importancia en lo que las propiedades moleculares se refiere, puede
asignarse inequívocamente a través del proceso de
difracción en los cristales (sin necesidad de recurrir a
patrones), gracias a
la denominada dispersión anómala que algunos
átomos presentan frente a la radiación X y que
además ha dado pie a la aparición de una metodología
de resolución en el caso de las macromoléculas.
No parece dificil comprender el hecho de que los
enantiómeros de
una molécula tengan propiedades desiguales, ya que
en
definitiva se trata de moléculas diferentes, pero en el caso
de
la actividad biológica este aspecto resulta especialmente
llamativo.
 Las
moléculas enantiómeras que se representan en la
figura de
la izquierda fueron puestas en el mercado por una empresa
farmacéutica y, obviamente, éstas mostraron
propiedades
diferentes. Las
moléculas enantiómeras que se representan en la
figura de
la izquierda fueron puestas en el mercado por una empresa
farmacéutica y, obviamente, éstas mostraron
propiedades
diferentes.
Las propiedades de DARVON (Dextropropoxyphene Napsylate) pueden
consultarse a través de este enlace, mientras que
NOVRAD (Levopropoxyphene
Napsylate) fue
retirada del mercado. |
La señal experimental en
difracción que nos
permite esta
diferenciación estructural es consecuencia del
hecho de que el
factor de
dispersión atómico deja de comportarse
como un número real cuando
la frecuencia de los rayos X es
similar a la frecuencia natural de absorción
atómica. Véase también el
apartado
dedicado a la dispersión anómala.
En estas condiciones deja de cumplirse la ley de Friedel y
por lo tanto los factores de estructura, tales como |Fh,k,l | y |F-h,-k,-l |,
serán "algo diferentes". Por lo tanto, la
comparación de los
denominados estimadores de Bijvoet,
es decir
la comparación
de los cocientes entre factores de
estructura observados para tales parejas, frente a los correspondientes
cocientes entre factores de estructura calculados con uno de los dos
posibles modelos absolutos, deben de mantener idéntico sesgo:
es
decir, que si un determinado cociente entre valores observados es menor
de la unidad, también debe de ocurrir así para el
mismo
cociente entre factores calculados, y alternativamente al
revés, y todo ello para un elevado número de
cocientes.
En el caso de que los cocientes entre valores observados tenga un
comportamiento contrario a los cocientes entre valores calculados,
sería señal inequívoca de que nuestro
modelo
absoluto es el erróneo, debiendo ser modificado de manera
coherente
.
El lector aventajado puede
también consultar
el contenido de este enlace sobre dispersión
anómala, preparado por Ethan A. Merritt.
RESULTADO FINAL
El conjunto de información que describe un modelo
cristalográfico final es muy concreto:
- Datos
sobre el experimento de difracción que nos condujo al
modelo:
longitud de onda de los rayos X y el patrón de
difracción
obtenido (la intensidad e indices hkl
de los miles, o hasta cientos de miles) de
las ondas dispersadas por el cristal,
- Dimensiones de la celdilla elemental
(calculadas a partir de la celdilla recíproca),
- Simetría del
cristal (también deducida de la simetría del
espacio recíproco), y
- Un
conjunto de datos que expresan la posición de cada
átomo
en la estructura, su estado de vibración térmica
y, en su
caso el denominado factor de ocupación (ó de
población), tal como muestra la siguiente tabla.
Las
posiciones atómicas suelen indicarse en forma de coordenadas
fraccionarias, es decir, en forma de "fracción de eje
cristalográfico" de la celdilla elemental, pero en
ocasiones,
especialmente en el campo de las macromoléculas, cuando se
quiere hacer mención exclusiva a la estructura molecular
aislada, ésta se suele indicar en forma de coordenadas
absolutas, es decir, expresadas en Angström y referidas a un
sistema de ejes ortogonales que poco tienen que ver con los ejes
cristalográficos.

Fragmento del
resultado cristalográfico de la estructura de una
proteína, expresado en el denominado formato "tipo PDB"
(Protein Data Bank), es
decir con coordenadas atómicas expresadas en
Angström y
referidas a un sistema de ejes ortogonales, diferentes de los
cristalogáficos.
Para mayor claridad de la tabla se han omitido las
estimaciones
de precisión de las coordenadas y de los restantes
datos
que afectan al átomo y que siempre forman parte del
resultado.
El factor de población
expresa la
"fracción
de
átomo" localizada en una posición concreta y
normalmente
este factor es la unidad. Pero el siginificado de este
parámetro requiere una aclaración para el
lector principiante, ya que se podría llegar a pensar que
existen
"fracciones de átomo", lo que carace de significado
físico. Habida cuenta de que las moléculas tienen
sus
movimientos, de que el experimento de difracción no es
instantáneo y que, por otra parte, los rayos X "ven" una
enorme
cantidad de celdillas elementales, parece razonable pensar que un
factor de población menor de la unidad representa el hecho
de
que, en una cierta cantidad significativa de celdillas, el
átomo
ha cambiado de posición, y por lo tanto el resultado
cristalográfico está "viendo", como promedio (en
tiempo y
celdilla), sólo una fracción del átomo
en la
posición dada por las correspondientes coordenadas.
Las coordenadas atómicas, y en general toda la
información extraída de un estudio
cristalográfico, se almacena en bases de datos accesibles,
algo
diferentes según el tipo de compuesto o molécula
y que
se comentarán en otro capítulo de estas
páginas.
REPRESENTACIONES GRÁFICAS DEL MODELO
 Del
modelo estructural final (coordenadas de los átomos, etc.)
se deriva directamente todo un conjunto de información
adicional que conduce al conocimiento detallado
de la estructura en sí, tales como son las distancias
interatómicas, ángulos de enlace,
ángulos de
torsión, planos moleculares, momento dipolar..., y en
general
cualquier detalle que pueda ser de utilidad para la
comprensión
de la funcionalidad y/o propiedades del material objeto del estudio,
sea éste de naturaleza molecular o no. Del
modelo estructural final (coordenadas de los átomos, etc.)
se deriva directamente todo un conjunto de información
adicional que conduce al conocimiento detallado
de la estructura en sí, tales como son las distancias
interatómicas, ángulos de enlace,
ángulos de
torsión, planos moleculares, momento dipolar..., y en
general
cualquier detalle que pueda ser de utilidad para la
comprensión
de la funcionalidad y/o propiedades del material objeto del estudio,
sea éste de naturaleza molecular o no.
Especialmente en el caso de moléculas biológicas
complejas, el uso de procesadores gráficos de alta calidad y
de
modelos relativamente simples, facilita mucho la comprensión
de
las relaciones entre estructura y funcionalidad, tal como se muestra en
la figura de la izquierda. |
Pero, además, las técnicas actuales de
representación gráfica permiten obtener
modelos
de una gran belleza y capacidad descriptiva para que el lector pueda
visualizar y comprender las estructuras, tal como demuestran algunos de
los ejemplos que se muestran a continuación:

 Izquierda: Modelo de
bolas y barras para representar
la estructura de un compuesto inorgánico simple
Derecha: Representación
para otro compuesto inorgánico, al cual se le ha
añadido
una representación poliédrica parcial
Izquierda: Modelo de
bolas y barras para representar
la estructura de un compuesto inorgánico simple
Derecha: Representación
para otro compuesto inorgánico, al cual se le ha
añadido
una representación poliédrica parcialIzquierda:
Modelo de bastones,
animado, para representar la estructura molecular y
empaquetamiento de un compuesto orgánico simple
Derecha: En
el caso de las moléculas biológicas, dada su
complejidad,
se suelen usar modelos simplificados, como éste de cintas
que
representa diferentes motivos estructurales típicos
(alfa-hélices, hebras-beta, etc.)


Izquierda:
Modelo
combinado de cintas y bastones para representar la
estructura dimérica de una proteína que
muestra un
ión sulfato en el interior representado mediante bolas
Derecha: Representación
de la superficie de una molécula biológica en
donde los colores
representan diferentes propiedades de hidrofobicidad. La flecha
representa el momento dipolar de la molécula
Finalmente,
haciendo uso de
información adicional de otras técnicas
(como la crio-microscopía electrónica), o
simplemente combinando dos modelos moleculares en diferentes
conformaciones, se pueden obtener modelos como los mostrados a
continuación. Más aún, utilizando los
tiempos de
exposición ultracortos de los rayos X producidos por los
láseres
de electrones libres,
los cristalógrafos son capaces de recopilar datos de
difracción de macromoléculas en diferentes
conformaciones, es decir, durante el transcurso de sus respectivas
tareas. De esta manera, utilizando una gran cantidad de
instantáneas de rayos X, podemos producir como una
película donde podemos seguir las modificaciones moleculares
y,
por lo tanto, comprender su función.


Izquierda:
Modelo
combinado entre la
estructura molecular de una proteína y la envolvente que
suministra la microscopía electrónica
de alta
resolución para mostrar un poro formado por la
asociación
de cuatro moléculas de proteína
Derecha: Modelo
simplificado que muestra los cambios estructurales de la
cadena principal de un enzima,
entre sus dos estados: activo (abierto) e inactivo (cerrado). Las dos
estructuras
límite (las de ambos estados) fueron determinadas mediante
cristalografía